Abstract
Lateral gene transfer (LGT) is essential to generate genomic recombinants between Chlamydia trachomatis strains to facilitate the evolution of the organism. Because there is no reliable laboratory-based gene transfer system for Chlamydia trachomatis Recombinants, in vitro generation of recombinants from antibiotic-resistant strains is being used to study LGT. However, the selection pressures imposed on recombinants in vitro are likely to affect the statistical properties of recombination relative to natural clinical recombinants, including prevalence at particular loci. We examined multiple loci for 16 in vitro-derived recombinants of ofloxacin- and rifampicin-resistant L1 and D strains, respectively, grown with both antibiotics, and compared them with the same sequenced loci among 11 clinical recombinants.
Breakpoints and recombination frequency were examined using phylogenetics, bioinformatics, and statistics. Clinical and in vitro isolates were neatly matched into two groups, without misclassification, using Ward’s minimum variance based on cutoff date. As expected, gyrA (confers resistance to ofloxacin) and rpoB (confers resistance to rifampicin) had significantly more breakpoints among in vitro recombinants than clinical recombinants (P < 0.0001 and P = 0.02). , respectively, using the Wilcoxon rank-sum test). Unexpectedly, trpA also had significantly more breakpoints for recombinants in vitro (P < 0.0001).
There was also a significant selection at other loci. The strongest bias was for ompA in strain D (P = 3.3 × 10-8). Our results indicate that the in vitro model differs statistically from natural recombination events. Additional genomic studies are needed to determine the factors responsible for the observed selection biases at unexpected loci and whether these are important for LGT to inform approaches to genetic manipulation of C. trachomatis.
Materials And Methods
- Reference strains and clinical isolates of C. trachomatis.
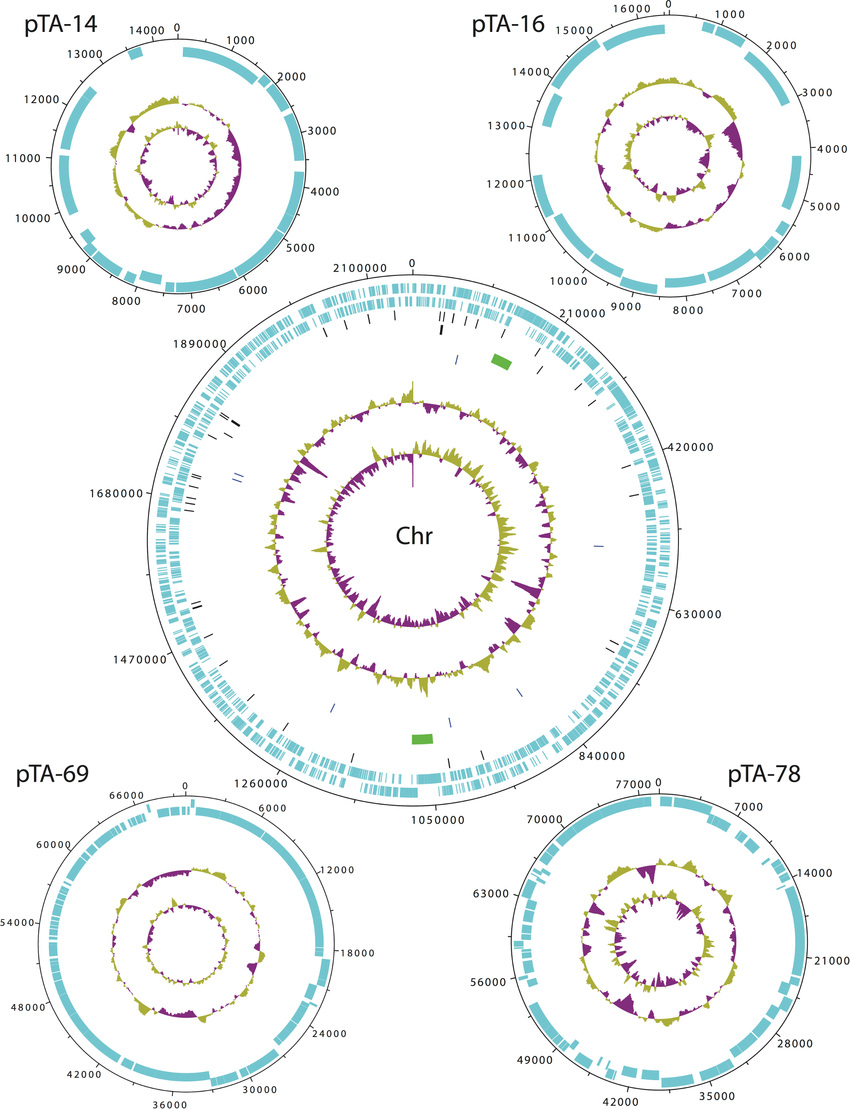
The following 16 reference strains of C. trachomatis were used in this study: A/HAR-13, B/TW-5, D/UW-3, Da/TW-448, E/Bour, F/IC-Cal3, G/UW-57, H/UW-4, I/UW-12, Ia/UW-20, J/UW-36, Ja/UW-92, K/UW-31, L1/440, L2/434 and L2b/UCH-1/proctitis. In addition, 11 urogenital samples were obtained from patients visiting sexually transmitted disease clinics. These samples represent ompA genotypes D, F, J, Ja, L2 and L2band were identified as recombinant by multilocus sequence typing (MLST) or whole-genome sequencing.
Briefly, both the reference and clinical strains were propagated in HeLa 229 cells, and elementary bodies were harvested and purified from host cells by batch density centrifugation in Renografin as we described previously. A high purity PCR template preparation kit (Roche Diagnostics) was used to extract genomic DNA following the instructions provided by the manufacturer. The ompA genotype of the 16 reference strains and 11 clinical strains was confirmed by comparison with sequences in GenBank using BLAST.
- Molecular evolution and phylogenetic analysis.
For each of the in vitro and clinical recombinants, the percentage G+C content of the genetic loci was calculated using EditSeq software (DNASTAR, Madison, WI). The molecular evolution of the genes of interest was evaluated using the Nei-Gojobori method to estimate the ratio of non-synonymous to synonymous substitutions (dN/dS) as previously described. Using the p-distance model, values were normalized against the number of potential dN and dS sites; Confidence intervals of 95% were used to determine significant differences in the mean of dN and dS. One thousand bootstrap replicates were performed to calculate the standard error.
The evolutionary history of the genes of interest was reconstructed using a bootstrap test of neighbour-joining tree topologies generated by MEGA where trees were created using the Kimura 2-parameter model and protein-level reconstructions were performed created using the gamma rate. Phylogenetic networks were constructed using SplitsTree4 as described previously. The network estimates the phylogenetic relationship between the 16 reference strains and the 11 clinical strains and was drawn according to default settings. The network was created based on 1000 boot replicas.
- Determination of genomic crossover regions and breakpoints.
Mosaic structures of the clinical isolates were proposed based on Simplot analyses as described previously. Briefly, the query sequence was compared to two potential parent strains and one outgroup strain using a window size of 100 bp with a sliding step size of 10 bp to locate and determine breakpoints. Significant informative sites were determined using the maximal chi-square test. Each informative site supports a phylogenetic tree based on Kimura’s 2-parameter model with confidence levels for each branch of the tree calculated from 1000 bootstrap replicates.
For each putative recombinant locus, a P-value was calculated using Fisher’s exact test for the best-supported breakpoint region (ie, between informative sites) based on the number of informative sites. A Bonferroni multiple-factor test for the number of ways was conservatively applied to choose the observed number of cutpoints from the observed number of informative sites for the locus (this is a standard binomial coefficient). The resulting P-value is the probability of observing the given pattern of informative sites supporting the two different ancestors in the absence of recombination.
- In vitro and clinical breakpoint analysis.
To compare the pattern of recombination between clinical and in vitro sequences, a table of breakpoints was generated from the pooled clinical and in vitro data. First, the data for seven genes (recF, Inca, trpA, gyrA, rpoB, murA, and on) and the intervening regions (int) between each gene were arranged according to gene order in the genome. This analysis was performed using complete (clinical samples) and partial (in vitro) sequence data of the genes. Intermediate regions do not represent sequence data; they represent only breakpoints between genes.
Second, for each of the 27 samples, a value of 1 was entered if a recombination breakpoint was present within the gene and a value of 0 was entered if no breakpoint was observed. In each intervening region, if both genes before and after the region have the same strain, then a value of 0 is entered. Otherwise, a value of 1 is entered, indicating that a recombination breakpoint resides somewhere within the intermediate region.
To determine if the clinical and in vitro samples have different recombination patterns, cluster analysis was performed. The patterns are different if the 27 samples can be grouped into distinct clinical and in vitro groups based on the presence or absence of recombination breakpoints within each region. Before performing the cluster analysis, the binary data described above were converted into a Jaccard distance dissimilarity matrix.